Assoziierte Forschungsgruppe Heidel
Aktuelle Projekte
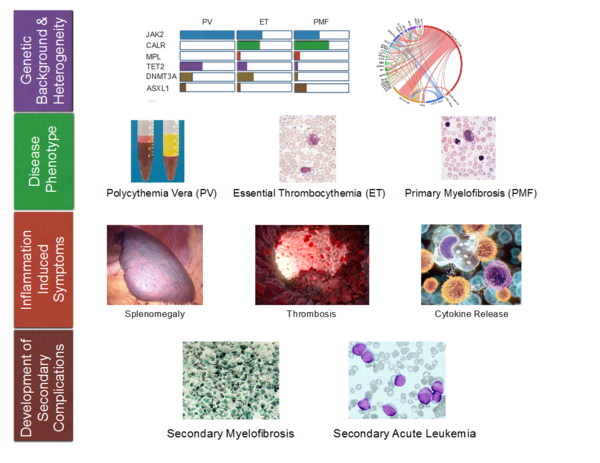
Bei der Philadelphia-Chromosom negativen myeloproliferativen Neoplasie (MPN) handelt es sich um eine klonale Erkrankung alternder hämatopoetischer Stammzellen sowie früher myeloischer Vorläuferzellen. Eine aktivierende Punktmutation der Janus Kinase 2 (JAK2V617F) steht im Mittelpunkt der Pathogenese der MPNs.
Diese Mutation findet sich bei etwa 95% der Patienten mit Polyzythämia vera (PV), sowie bei etwa 50% der Patienten mit essentieller Thrombozythämie (ET) oder Myelofibrose (MF). MPNs mit einer JAK2V617F-Mutation zeichnen sich durch eine vermehrte Proliferation einer oder mehrerer myeloischer Zellreihen aus sowie durch eine ausgeprägte systemische Entzündungsreaktion, vor allem im fortgeschrittenen Stadium der Erkrankung.
Die molekularen Mechanismen, die der JAK2V617F-induzierten Pathogenese zugrunde liegen, sind weitgehend unbekannt. Die therapeutischen Maßnahmen bei JAK2V617F-positiven MPNs sind bis dato auf eine symptomatische Behandlung der Patienten limitiert. Ziel der Arbeitsgruppe Heidel ist es, die molekularen Pathomechanismen dieser Erkrankung zu identifizieren und zu verstehen. Dazu führen die Forscher vergleichende in vitro und in vivo Untersuchungen zu den molekularen Prozessen durch, welche zur Entstehung und Erhaltung der Erkrankung beitragen. Ziel ist es letztendlich, neue Zielstrukturen zu identifizieren, um zukünftig die therapeutischen Optionen für die Patienten zu verbessern.
Ein Hauptmerkmal hämatopoetischer Stammzellen ist ihre Fähigkeit zur Selbsterneuerung. Verschiedene Gene und Signalwege kontrollieren dabei das sensible Gleichgewicht zwischen Selbst-Erneuerung und Differenzierung der hämatopoetischen Stammzellen, möglicherweise aber auch der leukämischen Stammzellen. Aktuelle Studien zeigen, dass sogenannte Determinanten des Zellschicksals wie z.B. RNA-bindende Proteine oder Polaritätsregulatoren die Biologie der Stammzellen beeinflussen können. Die Depletion der entsprechenden Gene mittels RNAi-Technologie führte zu einem verstärkten (Prox1) bzw. verringerten (Pard6a, Prkcz, Msi2) Repopulationspotenzial der Stammzellen in vivo.
Der Verlust der Zellpolarität hat durch eine veränderte Zell-Zell-Matrix-Interaktion Einfluss auf epitheliale Krebserkrankungen und fördert auch die Tumorentstehung. Außerdem ist die Regulation der Zellpolarität essentiell für die asymmetrische Zellteilung (“asymmetric cell division”, ACD). Diese wiederum wird benötigt, um festzulegen, ob eine Stammzelle bei ihrer Teilung entweder eine Stammzelle bleibt oder ausdifferenziert. Die asymetrische Zellteilung ist ein wichtiger Bestandteil der Zellpolarität und könnte einen essentiellen Einfluss sowohl auf die Hämatopoese als auch auf die Leukämogenese haben. ACD reguliert die Einleitung und Aufrechterhaltung der Polarität während der Zellteilung, was zur Entstehung zweier Tochterzellen mit unterschiedlichen genetischen Eigenschaften führt. Infolgedessen sind asymetrisch angeordnete Proteine oft Determinanten des Zellschicksals. Bei der normalen Hämatopoese ist die asymetrische Zellteilung für die Erhaltung des hämatopoetischen Stammzell-Pools verantwortlich. Teilt sich eine Stammzelle, dann durchläuft eine der Tochterzellen ein genetisches Program, welches Proliferations- und Differenzierungsprozesse induziert. Die zweite Tochterzelle wird durch ein Programm gesteuert, welches die Zelle stilllegt und ihr damit die Kapazität zur Persistenz verleiht. Die asymetrische Zellteilung bewirkt somit in hämatopoetischen Stammzellen eine asymetrische Segregation der Selbsterneuerung an eine der beiden Tochterzellen. Bei der Entstehung von Leukämien könnte eine gestörte ACD (z.B. durch Verlust von Proteinen, die an der Zellpolarität beteiligt sind,) sowohl zur Vererbung der Selbst-Erneuerung als auch der Proliferationskapazität führen und somit zur leukämischen Transformation.
Unterstützt man die Hypothese, das sich eine Leukämie sowohl aus einer hämatopoetischen Stammzelle als auch aus einer mehr ausgereiften Vorläuferzelle entwickeln kann, so könnte die ACD die Proliferationskapazität einer prä-leukämischen Stammzellpopulation oder umgekehrt, die Selbsterneuerungskapazität von weiter ausgereiften prä-leukämischen Progenitorzellen (“granulozyte-macrophage progenitors”, GMP) verstärken. Folglich würde der Verlust der ACD zu einem Ungleichgewicht zwischen der Expansion differenzierter Progenitorzellen und der Erhaltung der hämatopoetischen Stammzellen führen und somit zur Entstehung einer Leukämie beitragen.
Mit Hilfe genetisch veränderter “Knockout”-Mausmodelle untersuchen wir in vivo verschiedene Signalwege und Determinanten des Zellschicksals, welche die Selbst-Erneuerungskapazität beeinflussen.
Die Phosphatidylinositol-spezifische Phospholipase C (PLC) gehört zu einer Gruppe von Enzymen, die die Hydrolyse von Phosphatidylinositol-4,5-biphosphat katalysiert. Durch diesen Prozess entstehen die sekundären Botenmoleküle Inositol-1,4,5-triphosphat (IP3) und Diacylglycerol (DAG). IP3 führt zur Mobilisierung von intrazellulärem Kalzium und bewirkt somit eine Erhöhung der intrazellulären Konzentration an freien Kalzium-Ionen. DAG hingegen fungiert als endogener Aktivator der Proteinkinase C.
PLC gamma 1 (PLCg1) ist eine von zwei PLC gamma-Isoformen, welche durch Rezeptor- und Nichtrezptor-Tyrosinkinasen aktiviert werden können. PLCg1 agiert “downstream” von verschiedenen Membran-Rezeptoren (wie z.B. dem T-Zell-Rezeptor) sowie nicht Membran-gebundenen Kinasen wie JAK2 oder BCR-ABL
Wir vermuten, dass der PLCg1-Signalweg eine entscheidende Rolle für die Proliferationskapazität hämatopoetischer Stammzellen in vitro und in vivo spielt.
Unter Einsatz der RNAi-Technologie (shRNA “Knockdown”) sowie eines konditionalen PLCg1 “Knockout”-Mausmodells, möchten wir die funktionelle Relevanz dieses Signalweges in hämatopoetischen Stammzellen untersuchen. Dabei möchten wir zum einen den Einfluss von PLCg1 auf die Differenzierung, Proliferation und Selbsterneuerungskapazität der Stammzellen während des Alterns untersuchen und zum anderen die Rolle von PLCg1 in der Entwicklung myeloischer Neoplasien verstehen.
Kontakt

Florian H. Heidel
Assoziierter Gruppenleiter
+49 3641 65-6011
haematologie.onkologie@mh-hannover.de
Beate Gaulke
Sekretariat (Medizinische Hochschule Hannover)
+49 511 532-3020/3021